1078
Views & Citations78
Likes & Shares
Lung cancer, predominately non-small cell lung cancer (NSCLC), is the leading cause of deaths among all cancers worldwide, both in men and women. Advances on high-throughput sequencing facilitate development of molecular treatments targeting onco-driver genes with somatic mutations. Among these, epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) were developed to inhibit mutation-induced abnormal cell growth and proliferation with favorable outcomes in patients with specific EGFR mutations and are approved by FDA as first-line treatment for NSCLC. However, the diverse responsiveness of EGFR-TKI among different mutation types as well as the emerging drug resistance warrants further investigations of the distinct regulatory mechanisms of TKI in each EGFR mutation type. The mass-spectrometry (MS)-based proteomics technology is a powerful tool to systematically elucidate changes in the proteome expression level as well as the dynamic intracellular signaling in different biological systems. Various proteomics strategies have been utilized for characterization of signaling pathways related to EGFR mutations as well as identification of aberrant phosphorylation and proteins for predicting sensitivity to treatment. In this review, we discussed about recent updates on proteomic researches in NSCLCs with EGFR-TKI treatments, chiefly on TKI triggered phosphoproteomics and proteomics profiles and the discoveries of specific biomarkers related to drug sensitivity and resistance.
Keywords: Lung cancer; EGFR mutation; EGFR-TKI; Proteomics
INTRODUCTION
Malignancies in the respiratory tract and lung are the leading cause of cancer-related deaths for both men and women with a five-year survival rate ranging from 2% to 30% [1-3]. In 2018, at least 250,000 new cases are estimated to occur in the United States [4] and it is estimated that the mortality case will grow up to 3 million in 2035 [2]. The major histological type of lung cancer is non-small cell lung carcinoma (NSCLC) which accounts for approximately 85% of the lung cancer cases and comprises subtypes including adenocarcinoma which is frequently found in females (23%~68%), squamous cell carcinoma, commonly found in males (27%~55%) and fewer cases of large cell carcinoma [5]. Previously, measures to treat lung cancer will include surgery for earlier-stage patients, and/or combination of chemo- and radiotherapy. Due to the recent advances in high-throughput sequencing of cancer genome, several oncogenic genes with somatic mutations have been characterized to lead to the development of cancers.
Based on the genetic alternations, molecular treatments that would inhibit the induced abnormal signaling from somatic mutations were developed, including epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) which have been approved as a standard treatment for NSCLC with EGFR mutations, and also inhibitors of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), KRAS proto- oncogene, GTP ase (KRAS), and B - Raf proto-oncogene, serine/threonine kinase (BRAF).
Randomized trials comparing TKIs with platinum-based chemotherapy showed favorable outcome for TKIs in extending progression-free survival (PFS) (9-13 vs. 5-7 months) with response rates of approximately 70% in patients with known specific mutations [6-10].
Epidermal growth factor receptor (EGFR), a member of ErbB family of receptors, is the most predominant oncogenic driver in lung cancer. In physiologic condition, ligand binding triggers EGFR signaling by causing dimerization of the extracellular domain of EGFR and ATP-driven auto-phosphorylation in the kinase domain which subsequently activate complex signaling cascades in RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, PLCgamma-PKC or JAK/STAT pathways to promote cell growth, proliferation, and migration capacity, as well as activation of pro-survival signals. The occurrence of somatic mutations in EGFR would lead to a ligand- independent signaling pathway and exacerbate lung cancer development characterized by uncontrollable cell proliferation, increased angiogenic and metastatic capacity, and resistance to apoptotic signals [11-13]. Forty different mutations have been reported in EGFR in the Unipart knowledgebase of which mutations in the kinase domain, most commonly a point mutation in exon 21 (L858R) or deletions in exon 19 (e.g. deletion of amino acid E746 to A750), are present in at least 10% of Caucasian patients with adenocarcinoma subtype [14]. Asian patients even bear higher frequency of EGFR mutations (up to 60%), especially in female (20-58%) and in never- smoker patients (35-60%) [14-16]. Targeting on these mutations, the first (erlotinib and gefitinib), second (afatinib) and third (osimertinib) generation EGFR-TKIs are developed and approved by FDA for initial treatment of EGFR-mutated lung adenocarcinoma. Both erlotinib and gefitinib would reversibly compete for the ATP binding sites of EGFR and block activation of downstream signaling and exhibit response rate of 70% with significant improvement in overall survival to be beyond 20 months [10,17-20]. Afatinib acts by irreversibly binding to the tyrosine kinase of EGFR with higher potent inhibition compared to the first generations. The third generation osimertinib shows a significantly greater selectivity (up to 200 times) to EGFR mutant cells compared to the wild-type EGFR [21,22] and reduced adverse events compared to the first and second generations of EGFR-TKIs [23].
While these targeted therapies show promising improvement in patient’s PFS and overall survival, later studies have addressed variations of TKI responsiveness in different EGFR mutation types. For example, NSCLC cells expressing EGFR L858R mutation are more sensitive to gefitinib than are those that express G719S mutant [24]. NSCLC patients with exon 19 mutations showed higher response rate (70~100%) as well as longer overall survival of 26~36 months compared to the response rate of patients harboring exon 21 mutations (20-67%), who showed overall survival ranging from 8~17 months [25-29]. A group of 3 to 14% of the EGFR-mutant patients was diagnosed with multiple mutations within a single tumor sample and exhibited the poorest treatment response with PFS of less than 2 months [30,31]. In addition, almost all patients with initial response to EGFR-TKIs developed resistance to the drugs in within 6~12 months and subsequently underwent tumor progression [32,33]. Several mechanisms of acquired EGFR-TKI resistance have been proposed, including the acquisition of a secondary mutation at the gatekeeper residue (EGFR-T790M) which accounts for about 60% of the resistance [34]. Other mutations or alternations in the EGFR-downstream signaling molecules including amplification of MET/HGF, HER2 mutations, overexpression of HER3, persistent activation of IGF-1R, mutations of PIK3CA/AKT, loss or down regulation of PTEN, and abnormal dimerization of STAT 3 have also been reported to contribute to EGFR-TKI resistance [35,36]. Furthermore, in approximately 25% of the resistance cases the exact mechanism is still unknown. The huge variations in EGFR-TKI responses highlight the clinical needs to study the alterations in regulatory mechanism of EGFR-TKI for identification of potential biomarkers to predict EGFR-TKI response and discover alternative targets to overcome drug resistance.
As the effector of cellular function, studies in protein and their post-translation modifications are essential to investigate the spatial and temporal regulatory mechanism that are not able to be predicted by using genomics. Recent advances in the mass-spectrometry (MS)-based proteomics technologies have allowed us to systematically elucidate changes in the proteome expression levels as well as the dynamic intracellular signaling in various sample types including cells, tissues, or organisms for biomarker discovery. This large-scale analysis show promises in advancing the goal of “precision medicine”, which aims to distinguish patients on the basis of molecular characteristics for personalized treatments [37]. In this review, we will discuss about recent updates on proteomic researches in NSCLCs with EGFR-TKI treatments, chiefly on TKI triggered signaling pathways and the discoveries of specific biomarkers related to drug sensitivity and resistance.
Proteomics Strategies for Studying EGFR Signaling
A majority of proteomics focus on the characterization of the peptides digested from proteins by using tandem MS which refer to as bottom-up proteomics. Among these, shotgun or large-scale proteomics are most commonly used strategies to achieve a comprehensive identification of peptides and proteins in a data-dependent acquisition mode. Typically, the total cell lysate or subcellular protein fractions are extracted from cells, tissues or body fluid specimens using combinations of mechanical and chemical methods and digested into peptides using a proteolytic enzyme such as trypsin. The peptides are further separated into different fractions based on their physical properties using liquid chromatography (LC), including strong cat-ion exchange (SCX), anion exchange, and reversed phase (RP) chromatography. Each peptide fraction is submitted for RPLC-MS/MS analysis to acquire the MS/MS spectra for peptides with higher intensities. The MS/MS spectra are searched against target protein sequence database and decoy database using search engines, such as Mascot [38], Sequest [39], Protein Pilot [40] and Max Quant [41,42], to obtain the confident peptide and protein identifications.
In combination with either label-free or isotopic labeling techniques, the quantitative information of proteome can be obtained by measuring the MS signals of individual peptides in different samples (label-free quantitation) or the signals from isotopically labeled peptides in the same LC-MS analysis (isotopic labeling quantitation). Label-free quantification involves direct measurement of abundance of peptides from the analyzed sample by spectral counting (number of MS/MS spectra) or peak intensities (peak area or height) [43]. In isotopic labeling strategies, proteins or peptides are labeled with stable isotopes prior to MS analysis. Stable isotope labeling with amino acids in cell culture (SILAC) involves culturing cells with dialyzed medium containing amino acids with different isotopes (“heavy” 13C- or 15N-labeled arginine or lysine) to metabolically incorporate heavy amino acids into every protein. After the cell culturing, the heavy and light SILAC cells can be mixed in equal mount for further proteome analysis [44]. The quantitation of peptides is achieved based on the extracted peak area of the differentially labeled peptides during MS scanning [45]. Multiplexed isobaric labeling methods are used for peptide level labeling with tags that consist of identical overall mass but different distribution of heavy isotopes in the reporter and balance groups. These include isobaric tags for relative and absolute quantitation (ITRAQ) and tandem mass tags (TMT) [46]. Multiple samples (at most 10) are digested separately, labeled with different forms of isobaric tags, and then combined for LC-MS/MS analysis. During the MS/MS scan, the peptides with isobaric tags will be fragmented to produce reporter ions with different m/z which intensities can be used for peptide and protein quantitation [46]. For verification and validation of identified proteins, multiple reaction monitoring (MRM)-MS is applied to due to its high specificity and accuracy which monitors the pre-defined peptide precursor and fragment ions (transitions) in the triple quadruple MS. Absolute quantitation is obtained on the measurement of the peak areas for transitions which is interpolated into a calibration curve constructed using standard peptides [47].
Regarding the analysis of protein post-translational modifications, in particular phosphorylation for signal transduction, additional enrichment of phosphorylated peptides prior to MS is required because of the low abundance and low ionization efficiency of phosphorylated peptides compared to other unmodified peptides. For global phospho-proteomics analysis, immobilized metal affinity chromatography (IMAC) or titanium oxide (TiO2) are commonly applied to enrich phosphopeptides based on the interaction of the negatively charged phosphate groups and positively charged resin bed containing metal ions such as Fe3+, Al3+ and Ti4+ [48-50]. In addition, immunoprecipitation is used to selectively enrich phospho-tyrosine (pY) peptides by using specific antibodies [51]. Both of the enriched phosphopeptides and the flow through (proteome) could be analyzed to quantitatively compare the expression changes of site-specific phosphorylations and the proteins in the same experiment.
Elucidation of EGFR Signaling Cascades upon Treatment of TKIs by Using Quantitative Proteomics and Phospho-Proteomics
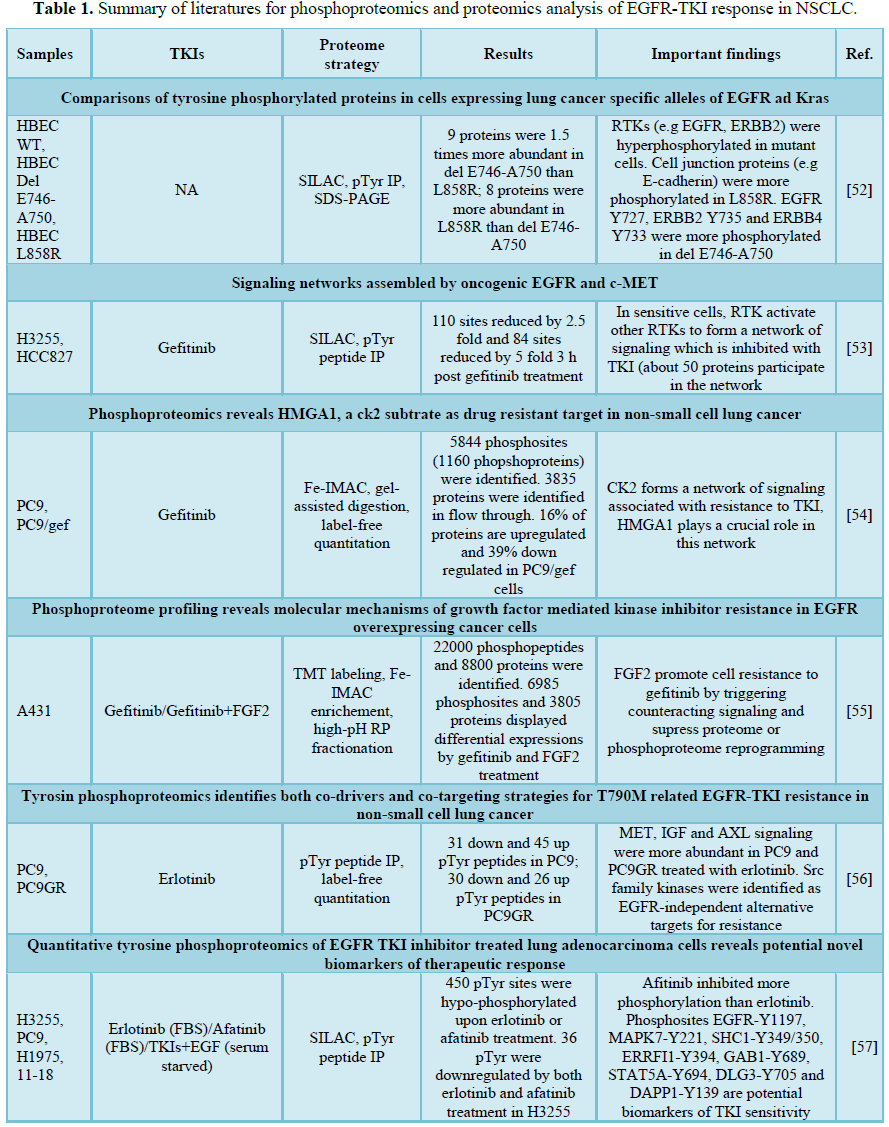
As shown in Table 1, several large-scale quantitative profiling as well as targeted quantitation of phosphoproteomes on lung adenocarcinoma cell lines with varying sensitivity to TKIs have been performed to provide a systematic molecular view of drug action and to elucidate the perturbed EGFR signaling upon treatment of TKIs. Considering the transcription to translation variations of different oncogenic mutations, Guha et al. [52] compared the baseline tyrosine phosphorylations in isogenic cells expressing lung cancer-specific alleles of EGFR (L858R and Del E746-A750) by using immune precipitation and SILAC-based quantitative proteomics analysis. Without TKI treatment, the level of tyrosine phosphorylation, especially on the tyrosine kinase receptors, was higher in EGFR mutant compared to those in wild-type (WT). A list of cell junction proteins showed more phosphorylated in cells expressing L858R than cells expressing Del E746-A750 [52]. To study the effect of gefitinib on downstream signaling, Guo et al. [53] performed phosphor-scan immune affinity enrichment and SILAC quantitation to the gefitinib-treated lung cancer cell lines, including sensitive EGFR mutant H3255 (L858R) and HCC827 (Del E746-A750) and identified dramatic decrease of tyrosine phosphorylations in sensitive mutant cells at EGFR signaling as well as at cell membrane and cell-cell junctions after 3 h and 24 h treatments. HER2, HER3, c-MET, MAGUK family proteins as well as other proteins not previously associated with EGFR signaling all showed significant de-phosphorylation upon drug treatment [53].
A continuous exposure of EGFR-TKI in sensitive EGFR-mutant NSCLC cells may induce drug resistance without induction of other mutations. To study the drug resistant mechanism, Wang et al. [54] performed phosphoproteomics and proteomics analysis using Fe-IMAC and label-free quantitation on a pair of gefitinib-treated TKI-sensitive PC9 and resistant PC 9/gef cells which were derived from continuous exposure of an increased concentration of gefitinib. A total of 3834 proteins and 4612 phosphopeptides corresponding to 1548 phosphoproteins were identified and revealed increased expressions of CK2-mediated network of which HMGA1, a substrate of CK2, was validated as a potential drug-resistant target in NSCLC [54]. On the other hand, Koch and colleagues studied the resistant mechanism in EGFR wild-type (WT) A431 cells treated by FGF2 and gefitinib by Fe-IMAC and TMT-based proteome analysis. Among 22,000 phosphopeptides, 571 and 2105 phosphosites were uniquely inhibited and upregulated by gefitinib treatment, respectively, which included kinases and phosphatases involved in RNA processing and cytoskeleton organization [55].
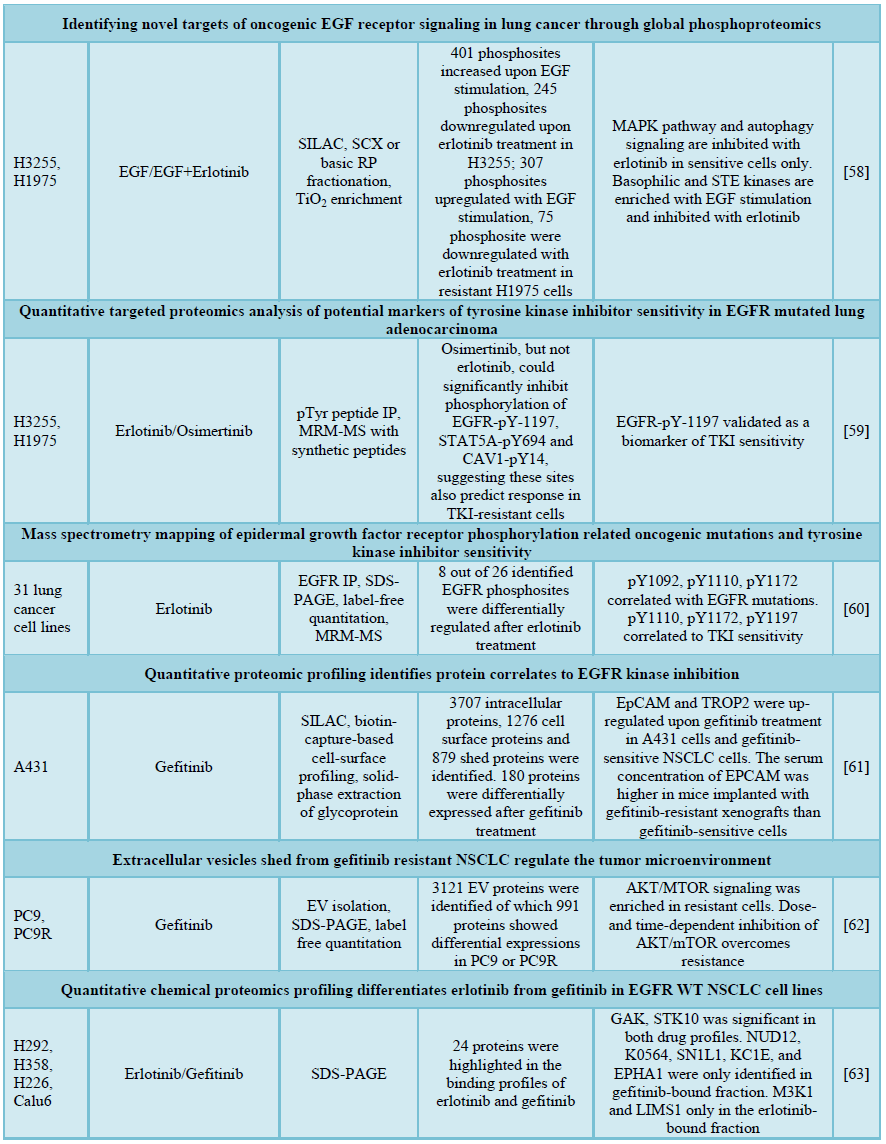
Regarding the mutation-induced erlotinib resistance, Yoshida and co-workers studies T790 M-related EGFR-TKI resistance using the immune affinity purification of tyrosine phosphorylation and label-free quantitative proteomics analysis on erlotinib-sensitive PC9 (Del E746-A750) and -resistant PC9GR cells bearing T790M. 403 unique phosphotyrosine (pY) peptides were identified of which 110 and 77 pY peptides showed up- or down-regulated in PC9GR cells. After erlotinib treatment, numerous receptor tyrosine kinases (RTKs), including MET, ROR1, YES, FAK, BRK, ERBB2 and AXL, as well as an adaptor protein IRS2 showed hyperactive in both PC9 and PC9GR cells, suggesting these RTKs may cooperate in coordinate to promote the EGFR-TKI resistance. In addition, Src family kinases (SFKs) were identified as EGFR-independent resistant pathway and the use of afitinib and dasatinib which simultaneous targeting EGFR T790M and SFKs were confirmed to suppress downstream signaling and produce better anti-tumor effects [56]. Zhang et al. [57] investigated the tyrosine phosphoproteomics changes in EGFR L858R and L858R/T790M cells with varying sensitivity to erlotinib and afatinib using SILAC-based proteomics analysis. A list of pTyr sites, including EGFR-Y1197, MAPK7-Y221, SHC1-Y349/350, ERRFI1-Y394, GAB1-Y689, STAT5A-Y694, DLG3-Y705 and DAPP1-Y139, were identified as potential biomarkers of TKI sensitivity [57]. Zhang et al. [58] applied TiO2 enrichment and SILAC quantitation to profile the global phosphorylation changes in EGFR L858R and L858R/T790M cells by erlotinib and EGF treatment. and identified hypophosphorylation of EGFR, insulin receptor, hepatocyte growth factor, mTOR, JAK/STAT and elf4/p70S6k signaling in H3255 (L858R) but not H1975 (L858R/T790) cells upon erlotinib treatment, suggesting that these signaling pathways could be candidate targets for lung cancer cases with T790M secondary mutations. On the other hand, EGFR-Y11172, Y1197, SHC1-Y428, SOS1-S1134 and NCK1-S85 were inhibited by erlotinib in resistant cells and therefore were considered as off-target effects of erlotinib [58]. They further developed immune-MRM method to verify 12 pTry sites in 9 proteins in the same cells with erlotinib or osimertinib treatment and validated EGFR-pY-1197 as a biomarker of TKI sensitivity [59]. To map the EGFR phosphorylations in NSCLC, Zhang et al. [60] measured the 30 phosphorylation sites of EGFR in 31 NSCLC cell lines and identified EGFR pTyr1092, pTyr1110, pTyr1172 to be correlated with activating mutations while three sites (pY1110, pY1172, pY1197) correlated with erlotinib sensitivity.
Other than the aberrant phosphoproteomics upon EGFR-TKIs, several studies focused on the quantitative proteomics analysis to offer alternative protein markers for EGFR signaling. Kani et al. [61] conducted total cell proteome, cell surface proteome and secretome analyses to identify 3707 intracellular proteins, 1276 cell surface proteins and 879 proteins shed into culture medium in gefitinib-treated A431 cells. Among these, EpCAM and TROP2 expressions were correlated to gefitinib resistance and the serum concentration of EpCAM was higher in mice implanted with gefitinib-resistant xynographts than gefitinib-sensitive cells. Extracellular vesicles (EV) have also been investigated for EGFR-TKI resistant markers. EVs isolated from PC9 and PC9R pre-treated with gefitinib showed a dramatic enrichment of proteins involved in the adherence junction, tight junctions, insulin and mTOR signaling including most prominently, PDK1, mTOR, AKT1 and AKT2 as involved in drugs resistance mechanism. In addition, dose- and time-dependent inhibition of AKT/mTOR pathways showed potential to overcome the resistance to gefitinib [62]. In order to evaluate different effectiveness of erlotinib and gefitinib in NSCLC patient with different EGFR genotypes, Augustin et al. [63] applied chemical proteomics analysis to find out the binding proteins and affinity of the two drugs using EGFR WT cell lines (H292, H322 and H226 and Calu6). The findings elucidated a substantially higher binding affinity of erlotinib for M3K1 and LIMS1 as compared to gefitinib. NUD12, K0564, SN1L1, KC1E, and EPHA1 were only identified in gefitinib-bound fraction. These results showed that multiple downstream signaling and protein interaction networks of EGFR-mutant cell lines were regulated in response to treatments and have contributed for the identification of sensitivity and resistance pattern of EGFR TKIs.
Identification of Serum Proteome Signatures for Predicting Response to TKI Treatments
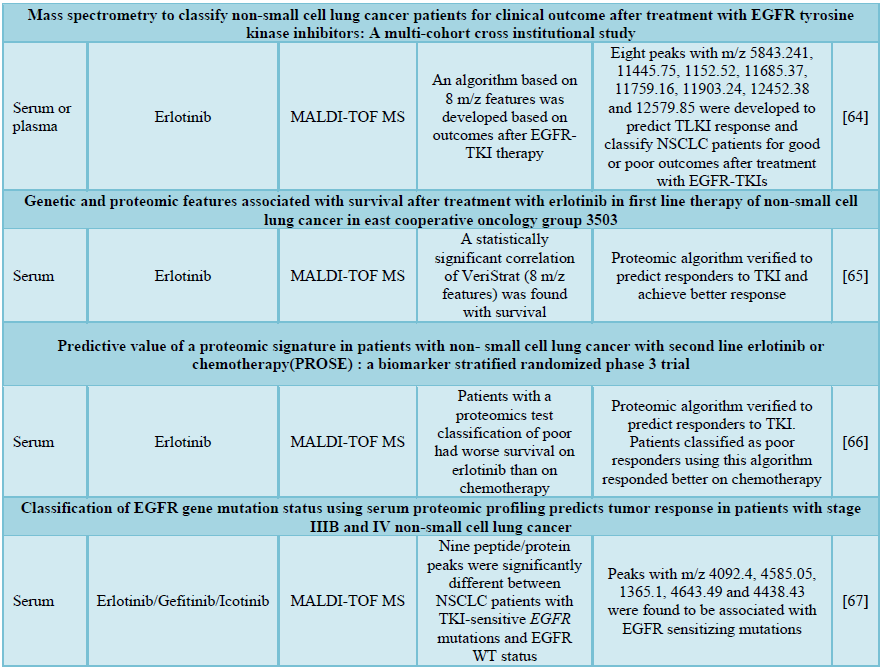
For clinical diagnosis and prognosis, non-invasive or slightly invasive detections of biomarkers in serum or plasma are very useful in the hospital setting due to convenience and minimal skill requirement. For this purpose, Taguchi et al. [64] conducted MALDI-TOF MS analysis on patient serum/plasma samples to characterize specific spectral features that can be applied as predictive markers for TKI in the clinic. By analyzing features of serum samples in multiple cohorts of NSCLC patients with stage IIIB or IV and comparing to their response upon erlotinib treatment, a classifier algorithm was developed based on eight peaks with m/z 5843.241, 11445.75, 1152.52, 11685.37, 11759.16, 11903.24, 12452.38 and 12579.85, to group patients into good and poor responders without taking into account of the EGFR mutation status. This algorithm achieved an improvement in the prediction of survival of patients that were administered TKI therapy [64]. Later, a follow-up study was conducted in which that both patients with WT and mutant EGFR status were recruited into the study. The proteomic classifier was consistently proven to be an effective predictor of response as shown by the longer survival for those pre-defined as good responders to erlotinib. Contrary to the belief that EGFR-TKI is suitable for patients harboring EGFR mutations only, it was found out that even patients with WT EGFR status were able to respond to erlotinib positively based on the proteomic classifier algorithm [65]. Another randomized phase 3 trial was also conducted to validate the power of response prediction for erlotinib. Erlotinib was initiated based on the prediction of good responders by the proteomic classifier and those who were classified as poor responders were administered chemotherapy. In consistent with previous results, patients classified as poor responders had longer survival on chemotherapy while those classified as good responders had a long survival on erlotinib treatment [66].
In addition, MALDI-TOF MS proteomic profiling had also been applied to investigate features that correlate with the presence of EGFR mutations. Peaks with m/z 4092.4, 4585.05, 1365.1, 4643.49 and 4438.43 were found to be significantly associated with EGFR sensitizing mutations. The proteomic classifier exhibited a sensitivity of 85.6% and a specificity of 77.5% in predicting patient EGFR mutation status. Treatment of patients by selective administration of TKI based on the proteomic classifiers rendered an objective response rate of 59.6% for patients with mutant EGFR while an objective response rate of 8.8% for patients with WT EGFR (p < 0.0001) [67]. These results are consistent with phosphoproteomics results which suggested that EGFR mutations are not sufficient for prediction of response to TKI but rather the signaling patterns driven by differential phosphorylation/protein expressions between patients provides more confident prediction.
Impacts of EGFR-TKIs in Site-Specific Phosphorylation Changes of EGFR-Related Signaling Pathways
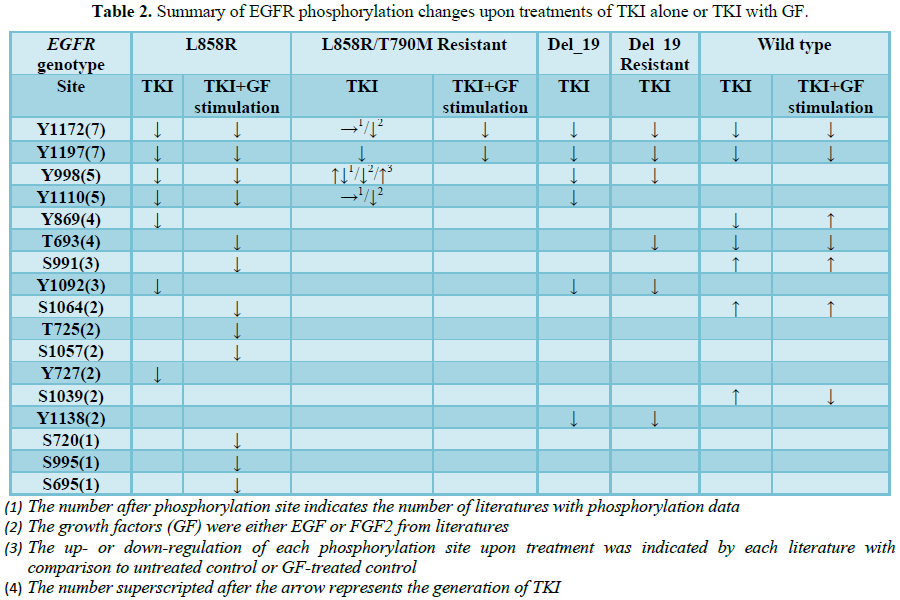
Based on the collected literatures, we further summarized the site-specific phosphorylation changes in EGFR upon treatments of TKI alone or TKI with growth factor (GF) treatment (Table 2). Upon TKI treatment, most of the sites in the cytoplasmic domain of EGFR showed general hypophosphorylations in both EGFR L858R and exon 19 deletion mutated cell lines. In addition, EGFR WT cells also showed similar hypophosphorylation pattern of several sites, including Y1172, Y1197, T693 and Y869, while S991, S1064, and S1039 had upregulated phosphorylation. In TKI-resistant mutant cell lines, varied phosphorylation responses to EGFR-TKI have been noticed. In L858R/T790M resistant cell lines, Y1172 and Y1110 phosphorylations remained unchanged upon first generation-TKI treatment, and it were hypophosphorylated upon second generation-TKI treatment [53,56-58]. Also, Y998 showed inconsistent findings upon first generation-TKI treatment, while it showed downregulated and upregulated phosphorylations upon second and third-generation TKI treatment, respectively [53,56,57,59]. In resistant exon 19 deletion cell lines, these variations were less observed, possibly due to the higher response rate of patients with Del E746-A750 mutation to TKIs.
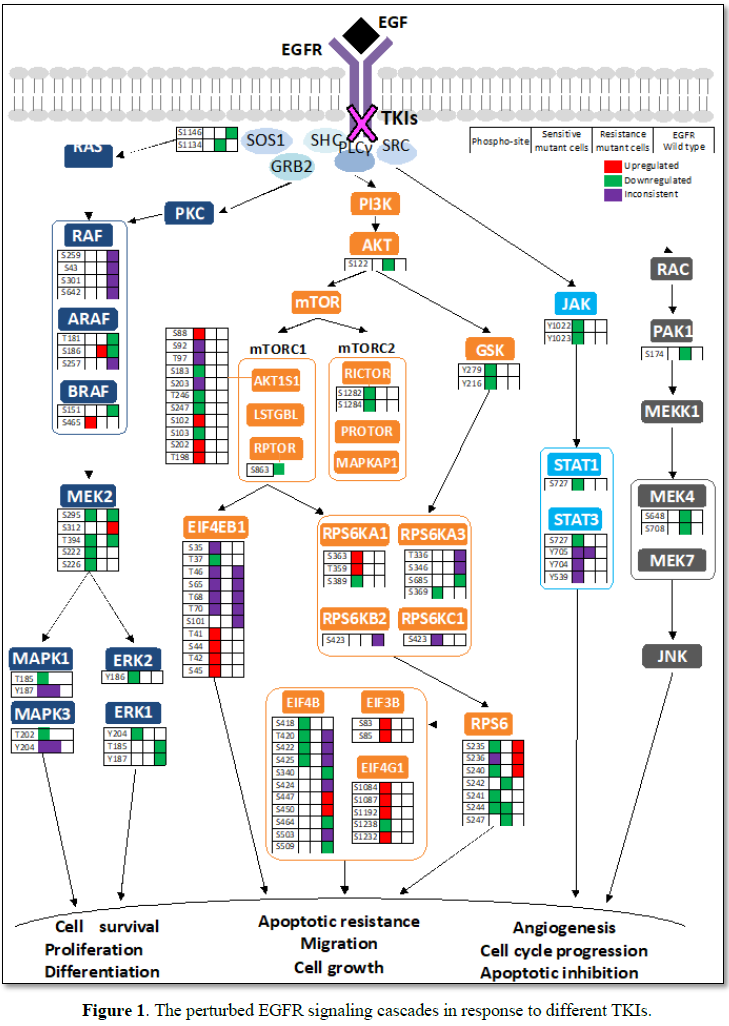
Analysis of global hypophosphorylation upon EGFR-TKI treatments has revealed a wide range of proteins and pathway axes affected by the therapy (Figure 1). The majority of them are targeted of mutant EGFR but some of them are neither associated to EGFR nor its mutation induced-signaling. Multiple downstream signaling pathways of EGFR activation showed alterations upon EGFR-TKI treatment. In L858R mutated cell lines, MEK2 and MAPK in RAS/RAF pathway showed hypophosphorylation both in basal level and upon TKI treatment [57,58], although unchanged phosphorylation levels were also observed in MAPK13, MAPK14 and MAPK7 [57]. Upon TKI treatment, additional downregulation of phosphorylation level was observed in Y204, Y185 and Y220 sites of ERK1, ERK2 and ERK5, respectively, indicating potential targets that were also regulated by TKIs [53]. In L858R resistant cells, interestingly, phosphorylation sites of RAF1, BRAF, MEK2 and MAPK complexes showed no alterations upon TKI and GF stimulation [57,58], with noted increase in S186 phosphorylation of ARAF [55]. Furthermore, treatment with second-generation TKI showed potential to induce hypophosphorylation of MAPK complex (shown in green in Figure 1). Similarly, while AKT1 and GSK3A in the PI3K/AKT/MTOR pathway seemed not to be affected by EGFR mutation, some phosphorylation sites of mTOR complexes, including RICTOR, AKT1S1, RPTOR, RPS6 and also EIF4B, the effector that control cell cycle, showed hypophosphorylation upon TKI-GF treatment [55,58]. However, these phosphorylation changes were not detected in L858R resistant cell lines [58].
It is also crucial to note that Del E746-A750 mutated cells showed distinct regulation of these downstream signaling pathways. Upon TKI treatment, sites on AKT1S1 showed inconsistent phosphorylation pattern, with some underwent hypophosphorylation (S92, S203, T246 and S103) and some underwent hyperphosphorylation (T97, S102, S202 and T198). In addition, proteins of the mTOR complexes, as well as the EIF4 complex also showed hyperphosphorylation [55,58]. Del E746-A750 mutated cells, both sensitive and resistant types, exhibited hyperphosphorylation of STAT3 upon TKI treatment [53]. Wild-type cells also exhibited hypophosphorylation pattern in SOS1, RAF1, ARAF and MEK2 upon TKI treatments [55,58]. Altogether, these findings suggested that different EGFR-mutation type induces unique activation properties of the EGFR-downstream signaling pathways and TKI treatment resulted in distinct regulations of these pathways. Study of the pathways that are not enriched in resistant cells upon TKI treatment may provide a proper direction on how TKI resistance can be overcome.
FUTURE PERSPECTIVE
Despite of the discovery of oncogenic driver and regulatory gene mutations in cancers, the exact role of these mutations in cancer onset and progression remains unknown, and accurate prediction on which mutation will lead to cancer progression (driver mutation) or without risk of cancer development (passenger mutations) is still limited. Alterations in transcription and translation processes result in distinct phenotypical characteristics between diseases. Multiple clinical trials failed to show consistent response to EGFR-TKI treatment. Thus, we believe that genetic mutation studies solely in the EGFR gene are not ideal indicators of pathway activities. Combining EGFR genetic test and TKI triggered EGFR-related signaling cascades as potential biomarkers would provide better prediction for drug sensitivity and resistance and for searching alternative therapeutic targets.
In addition, some challenges remain to be overcome for clinical application. Based on current progress, most of the experiments were done based on cell line analysis which do not truly reflect the heterogeneous nature of tumor tissue and its microenvironment. However, because of the invasive approach for collecting patient tissue samples, only a low quantity of sample is available for proteome and phosphoproteome analysis which hampers both discovery and validation studies using patients’ tissues. Therefore, samples that require a fairly noninvasive method such as blood and fluid from broncho-alveolar lavage are promising alternatives that can be used in the clinic. However, there is a need for customizing the proteomics workflow to improve on overcoming the variations between the body fluid samples and tumor tissues. The collection and storage of the clinical samples are also critical to alter protein/phosphorylation expression changes [68,69]. With the accelerating developments in sample preparation and proteomics technologies, we believe that the MS-based workflows will be made more robust for clinical applications. Appropriate use of artificial intelligence is also another promising solution that can help in handling of large data and result interpretation to the point of assisting clinicians on the best decision to take.
ACKNOWLEDGMENT
This research was financially supported by the Ministry of Science and Technology of Taiwan (MOST106-2113-M-038-004-MY2) and Taipei Medical University (TMU103-AE1-B14) in Taiwan. AE1-B14) in Taiwan. Alterations in transcription and translation processes result in distinct phenotypical characteristics between diseases.
- Cheng TY, Cramb SM, Baade PD, Youlden DR, Nwogu C, et al. (2016) The international epidemiology of lung cancer: Latest trends, disparities and tumor characteristics. J Thorac Oncol 11: 1653-1671.
- Didkowska J, Wojciechowska U, Manczuk M, Lobaszewski J (2016) Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann Transl Med 4: 150.
- Wong MCS, Goggins WB, Yip BHK, Fung FDH, Leung C, et al. (2017) Incidence and mortality of kidney cancer: Temporal patterns and global trends in 39 countries. Sci Rep 7: 15698.
- Siegel RL, Miller KD, Jemal A (2018) Cancer statistics 2018. CA Cancer J Clin 68: 7-30.
- Youlden DR, Cramb SM, Baade PD (2008) The international epidemiology of lung cancer: Geographical distribution and secular trends. J Thorac Oncol 3: 819-831.
- Foster P, Yamaguchi K, Hsu PP, Qian F, Du X, et al. (2015) The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol Cancer Ther 14: 931-940.
- Shan L, Wang Z, Guo L, Sun H, Qiu T, et al. (2015) Concurrence of EGFR amplification and sensitizing mutations indicate a better survival benefit from EGFR-TKI therapy in lung adenocarcinoma patients. Lung Cancer 89: 337-342.
- Ling Y, Yang X, Li W, Li Z, Yang L, et al. (2016) Overexpression of mutant EGFR protein indicates a better survival benefit from EGFR-TKI therapy in non-small cell lung cancer. Oncotarget 7: 52862-52869.
- Wu TH, Hsiue EH, Lee JH, Lin CC, Liao WY, et al. (2018) Best response according to RECIST during first-line EGFR-TKI treatment predicts survival in EGFR mutation-positive non-small-cell lung cancer patients. Clin Lung Cancer 19: e361-e372.
- Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, et al. (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13: 239-246.
- Bunn PA Jr, Franklin W (2002), Epidermal growth factor receptor expression, signal pathway and inhibitors in non-small cell lung cancer. Semin Oncol 29: 38-44.
- Inamura K, Ninomiya H, Ishikawa Y, Matsubara O (2010) Is the epidermal growth factor receptor status in lung cancers reflected in clinicopathologic features? Arch Pathol Lab Med 134: 66-72.
- Zhang Z, Stiegler AL, Boggon TJ, Kobayashi S, Halmos B (2010) EGFR-mutated lung cancer: A paradigm of molecular oncology. Oncotarget 1: 497-514.
- Gazdar AF (2009) Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28: S24-31.
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, et al. (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97: 339-346.
- Kohno T, Nakaoku T, Tsuta K, Tsuchihara K, Matsumoto S, et al. (2015) Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res 4: 156-164.
- Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, et al. (2011) Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 29: 2866-2874.
- Zhou C, Wu YL, Chen G, Feng J, Liu XQ, et al. (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG- 0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12: 735-742.
- Wu YL, Fukuoka M, Mok TS, Saijo N, Thongprasert S, et al. (2013) Tumor response and health-related quality of life in clinically selected patients from Asia with advanced non-small-cell lung cancer treated with first-line gefitinib: Post hoc analyses from the IPASS study. Lung Cancer 81: 280-287.
- Gaughan EM, Costa DB (2011) Genotype-driven therapies for non-small cell lung cancer: Focus on EGFR, KRAS and ALK gene abnormalities. Ther Adv Med Oncol 3: 113-125.
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, et al. (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 4: 1046-1061.
- Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, et al. (2014) Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem 57: 8249-8267.
- Liao BC, Lin CC, Yang JC (2015) Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Curr Opin Oncol 27: 94-101.
- Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, et al. (2005) Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res 65: 8968-8974.
- Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, et al. (2016) Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun 7: 13665.
- Lievre A, Ouine B, Canet J, Cartier A, Amar Y, et al. (2017) Protein biomarkers predictive for response to anti-EGFR treatment in RAS wild-type metastatic colorectal carcinoma. Br J Cancer 117: 1819-1827.
- Mitsudomi T, Kosaka T, Endoh H, Horio Y, Hida T, et al. (2005) Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with post-operative recurrence. J Clin Oncol 23: 2513-2520.
- Jackman DM, Yeap BY, Sequist LV, Lindeman N, Holmes AJ, et al. (2006) Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res 12: 3908-3914.
- Riely GJ, Pao W, Pham D, Li AR, Rizvi N, et al. (2006) Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res 12: 839-844.
- Zhang B, Wang S, Qian J, Yang W, Qian F, et al. (2018) Complex epidermal growth factor receptor mutations and their responses to tyrosine kinase inhibitors in previously untreated advanced lung adenocarcinomas. Cancer 124: 2399-2406.
- Yang Y, Zhang B, Li R, Liu B, Wang L (2016) EGFR-tyrosine kinase inhibitor treatment in a patient with advanced non-small cell lung cancer and concurrent exon 19 and 21 EGFR mutations: A case report and review of the literature. Oncol Lett 11: 3546-3550.
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, et al. (2004) EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 304: 1497-1500.
- Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, et al. (2010) Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 28: 357-360.
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, et al. (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2: e73.
- Lin Y, Wang X, Jin H (2014) EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res 4: 411-435.
- Huang L, Fu L (2015) Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B 5: 390-401.
- Jameson JL, Longo DL (2015) Precision medicine - Personalized, problematic and promising. N Engl J Med 372: 2229-2234.
- Brosch M, Yu L, Hubbard T, Choudhary J (2009) Accurate and sensitive peptide identification with Mascot percolator. J Proteome Res 8: 3176-3181.
- Moore RE, Young MK, Lee TD (2002) Qscore: An algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom 13: 378-386.
- Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, et al. (2007) The paragon algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics 6: 1638-1655.
- Han X, Aslanian A, Yates JR 3rd (2008) Mass spectrometry for proteomics. Curr Opin Chem Biol 12: 483-490.
- Iwamoto N, Shimada T (2018) Recent advances in mass spectrometry-based approaches for proteomics and biologics: Great contribution for developing therapeutic antibodies. Pharmacol Ther 185: 147-154.
- Zvonok N, Xu W, Williams J, Janero DR, Krishnan SC, et al. (2010) Mass spectrometry-based GPCR proteomics: Comprehensive characterization of the human cannabinoid 1 receptor. J Proteome Res 9: 1746-1753.
- Chen X, Wei S, Ji Y, Guo X, Yang F (2015) Quantitative proteomics using SILAC: Principles, applications and developments. Proteomics 15: 3175-3192.
- Mann M (2006) Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol 7: 952-958.
- Rauniyar N, Yates JR 3rd (2014) Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res 13: 5293-5309.
- Cohen Freue GV, Borchers CH (2012) Multiple reaction monitoring (MRM): Principles and application to coronary artery disease. Circ Cardiovasc Genet 5: 378.
- Thingholm TE, Larsen MR (2009) The use of titanium dioxide micro-columns to selectively isolate phosphopeptides from proteolytic digests. Methods Mol Biol 527: 57-66.
- Dunn JD, Reid GE, Bruening ML (2010) Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom Rev 29: 29-54.
- Fila J, Honys D (2012) Enrichment techniques employed in phosphoproteomics. Amino Acids 43: 1025-1047.
- Breitkopf SB, Asara JM (2012) Determining in vivo phosphorylation sites using mass spectrometry. Curr Protoc Mol Biol 19: 1-27.
- Guha U, Chaerkady R, Marimuthu A, Patterson AS, Kashyap MK, et al. (2008) Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proc Natl Acad Sci U S A 105: 14112-14117.
- Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, et al. (2008) Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 105: 692-697.
- Wang YT, Pan SH, Tsai CF, Kuo TC, Hsu YL, et al. (2017), Phosphoproteomics reveals HMGA1, a CK2 substrate, as a drug-resistant target in non-small cell lung cancer. Sci Rep 7: 44021.
- Koch H, Wilhelm M, Ruprecht B, Beck S, Frejno M, et al. (2016) Phosphoproteome profiling reveals molecular mechanisms of growth-factor-mediated kinase inhibitor resistance in EGFR-overexpressing cancer cells. J Proteome Res 15: 4490-4504.
- Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, et al. (2014) Tyrosine phosphoproteomics identifies both co-drivers and co-targeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin Cancer Res 20: 4059-4074.
- Zhang X, Maity T, Kashyap MK, Bansal M, Venugopalan A, et al. (2017) Quantitative tyrosine phosphoproteomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor-treated lung adenocarcinoma cells reveals potential novel biomarkers of therapeutic response. Mol Cell Proteomics 16: 891-910.
- Zhang X, Belkina N, Jacob HK, Maity T, Biswas R, et al. (2015) Identifying novel targets of oncogenic EGF receptor signaling in lung cancer through global phosphoproteomics. Proteomics 15: 340-355.
- Awasthi S, Maity T, Oyler BL, Qi Y, Zhang X, et al. (2018) Quantitative targeted proteomic analysis of potential markers of tyrosine kinase inhibitor (TKI) sensitivity in EGFR mutated lung adenocarcinoma. J Proteomics pii: S1874-3919(18)30164-7.
- Zhang G, Fang B, Liu RZ, Lin H, Kinose F, et al. (2011) Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J Proteome Res 10: 305-319.
- Kani K, Faca VM, Hughes LD, Zhang W, Fang Q, et al. (2012) Quantitative proteomic profiling identifies protein correlates to EGFR kinase inhibition. Mol Cancer Ther 11: 1071-1081.
- Choi DY, You S, Jung JH, Lee JC, Rho JK, et al. (2014) Extracellular vesicles shed from gefitinib-resistant non-small cell lung cancer regulates the tumor microenvironment. Proteomics 14: 1845-1856.
- Augustin A, Lamerz J, Meistermann H, Golling S, Scheiblich S, et al. (2013) Quantitative chemical proteomics profiling differentiates erlotinib from gefitinib in EGFR wild-type non-small cell lung carcinoma cell lines. Mol Cancer Ther 12: 520-529.
- Taguchi F, Solomon B, Gregorc V, Roder H, Gray R, et al. (2007) Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: A multicohort cross-institutional study. J Natl Cancer Inst 99: 838-846.
- Amann JM, Lee JW, Roder H, Brahmer J, Gonzalez A, et al. (2010) Genetic and proteomic features associated with survival after treatment with erlotinib in first-line therapy of non-small cell lung cancer in Eastern Cooperative Oncology Group 3503. J Thorac Oncol 5: 169-178.
- Gregorc V, Novello S, Lazzari C, Barni S, Aieta M, et al. (2014) Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): A biomarker-stratified, randomised phase 3 trial. Lancet Oncol 15: 713-721.
- Yang L, Tang C, Xu B, Wang W, Li J, et al. (2015) Classification of epidermal growth factor receptor gene mutation status using serum proteomic profiling predicts tumor response in patients with stage IIIB or IV non-small-cell lung cancer. PLoS One 10: e0128970.
- Bugovsky S, Winkler W, Balika W, Allmaier G (2015) Long time storage (archiving) of peptide, protein and tryptic digest samples on disposable nano-coated polymer targets for MALDI MS. EuPA Open Proteomics 8: 48-54.
- Lee DH, Kim JW, Jeon SY, Park BK, Han BG (2010) Proteomic analysis of the effect of storage temperature on human serum. Ann Clin Lab Sci 40: 61-70.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Journal of Genomic Medicine and Pharmacogenomics (ISSN:2474-4670)
- Journal of Genetics and Cell Biology (ISSN:2639-3360)
- Journal of Astronomy and Space Research
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)
- Food and Nutrition-Current Research (ISSN:2638-1095)


